Some chemotherapies block cancer growth by driving tumour cells into a state of cell-division arrest termed senescence. It emerges that such cells have a boosted capacity to drive tumour growth if they exit senescence.
If cells encounter certain types of stress, they can enter a state of cell-division arrest termed senescence1, which is usually thought to be irreversible. Senescence protects organisms from potentially dangerous cellular proliferation, for example by preventing cell division after severe DNA damage. Many anti-cancer therapies cause cancer-cell senescence, which is considered to be a positive outcome of such treatment. However, in a paper online in Nature, Milanovic et al.2 reveal the unexpected twist that chemotherapy-induced senescence might generate tumour cells that have an enhanced potential to drive tumour growth if they exit senescence.
Senescence induction has been studied intensively for decades. The phenomenon was first described in fibroblast cells grown in vitro, and entry into the senescent state in this context was considered to be a hallmark of cellular ageing3. Subsequent research has revealed that the induction of senescence is a cellular response that occurs during both physiological and pathological processes1.
Senescence-associated reprogramming promotes cancer stemness
The protein p53 is one of the key proteins that can act as a cellular sensor and drive a cell to enter senescence. It responds to DNA damage, and its action can cause permanent cell-cycle arrest by activating the proteins p16INK4a and p21. A senescent state can also be promoted by addition of methyl groups to specific amino-acid residues on histone proteins that bind DNA4. This methylation results in chromosomal compaction, which keeps the DNA in a transcriptionally inactive conformation and thus helps to make entry into senescence irreversible.
It had been previously observed that some proteins that regulate the senescent state also have key functional roles in stem cells5. Milanovic and colleagues2 investigated whether there might be a stem-cell connection to senescence induced by chemotherapy. Analysing gene-expression profiles in mice in a type of cancer called lymphoma, the authors observed that cellular signalling pathways activated during chemotherapy-induced senescence are similar to the gene-expression patterns observed in stem cells, patterns that collectively define a cellular state called stemness.
Stem cells are at the top of the cell-division hierarchy and are thought to be able both to divide indefinitely and to generate the distinct cells present in a given tissue6. Moreover, stem cells have also been found within tumours, and experimental evidence indicates that cancer stem cells can drive cancer growth, as well as aiding tumour-cell migration and dispersal to other locations in the body in a process called metastasis4.
It seems counter-intuitive that the induction of senescence in cancer cells that arrests tumour growth would drive the gene-expression programs associated with the stem cells that drive the disease. Yet the authors consistently made this observation when they investigated a variety of cancer model systems of both human and mouse origin.
To investigate whether these acquired stemness features affect growth when cancer cells escape from senescence, Milanovic and colleagues used a genetically engineered tumour in mice in which a state of cell-cycle arrest could be maintained by administration of the drug tamoxifen (Fig. 1). Surprisingly, the authors observed that cells exiting senescence when tamoxifen was removed have a greater capacity to drive tumour growth than do control tumour cells that did not go through a senescent phase. The authors therefore conclude that senescence induction in cancer could have an unexpected ‘dark side’ if such tumour cells break through the cell-cycle-arrest barrier.
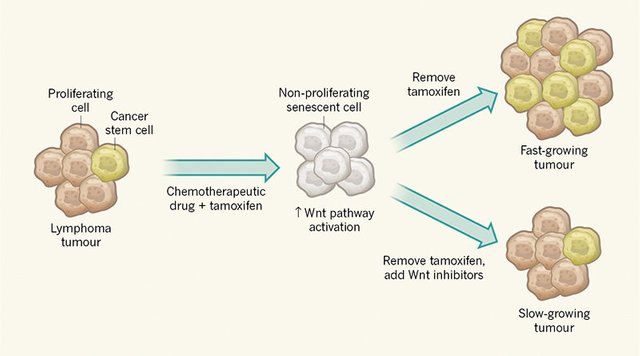
This is not the first indication that senescence might come at a cost. For example, senescent cells secrete a range of cytokine proteins that have a tumour-promoting effect on cancer cells in the vicinity by stimulating the stem-cell properties of such cells7,8. Milanovic and colleagues’ work, however, goes beyond observations of an indirect effect by revealing that senescent tumours have an intrinsic capacity to form an increased proportion of cancer stem cells. Although a role in this process for cytokines produced by other cells is not definitely excluded, the authors’ single-cell analysis is consistent with the phenomenon being cell-autonomous. This analysis reveals that cancer cells that have senescent hallmarks can, on release from senescence, proliferate and show hallmarks of cancer stem cells. The cancer-stem-cell features gained by these post-senescent cells cannot be explained by these cells simply being a cancer-cell subset that failed to enter senescence, because the authors show that entering senescence is a requirement for this process to occur.
The authors found a link between the activation of the Wnt signalling cascade and the senescent state. The observation that this well-studied stem-cell signalling pathway is activated during senescence provides additional confirmation of the surprising link with the induction of stem-cell characteristics. However, it is not clear why this pathway is activated. Nor is it clear whether Wnt ligands are secreted by senescent cells and whether such ligands then act on the same cell that secretes the protein or on neighbouring cells.
Notably, this finding also offers a means of targeting the potentially harmful effects of the cancer stem cells generated. The authors found that treatment of cells with a Wnt-pathway inhibitor could decrease tumour growth on exit from senescence. This discovery should be investigated in the clinic to determine whether it could enhance the effectiveness of chemotherapy.
Although these studies provide strong evidence for a close link between senescence and stemness, most of the work used a genetically engineered model system that allows exit from senescence to be controlled at will by removing a drug. How cancer cells might naturally break through senescence barriers in vivo, and whether this might be linked to acquisition of cancer stemness, should be investigated. The authors tried to address this by analysing spontaneous escape from senescence in samples of cancer cells from their mouse model grown in vitro, and also detected increased cancer stemness features in this context. Additional confirmation of these findings in non-genetically modified cancer models will, however, be needed. Nevertheless, Milanovic and colleagues’ data provide compelling evidence in the systems they studied that, when cancer cells escape from senescence, they have an enhanced capacity to drive tumour growth — a finding that has potential clinical implications.
This is a great post,this is ths first time i will see blog on cancer now i have more knowledge on it thanks for sharing.
Downvoting a post can decrease pending rewards and make it less visible. Common reasons:
Submit
itsok
Downvoting a post can decrease pending rewards and make it less visible. Common reasons:
Submit